



Medical Device News Update - October 2024
FDA approvals, warning letters and Class I recalls issued during October 2024

Monthly roundup of FDA approvals, warning letters and recalls issued in October 2024

Each month, Let’s Talk Risk! (LTR) provides a comprehensive summary of new device approvals or clearances, warning letters, and Class I recall announcements issued by the FDA in the previous month.
As a risk practitioner, it is very important to stay up to date with the latest regulatory developments affecting the medical device industry. A review of latest innovations approved or cleared by the FDA helps in understanding different ways the agency evaluates benefit-risk of medical devices when reviewing their safety and effectiveness. Warning letters and recall announcements are useful in building awareness of gaps in quality management system and the risk management process that should be addressed before they are cited by the FDA.
Only publicly available information is curated in this article. Links to relevant sources are provided in the footnotes.
Quick Summary
New device approvals/clearances
An ultrasonic propulsion device to facilitate removal of urinary stone fragments (De Novo DEN230082).
An electromechanical surgical system for tissue manipulation and repair during laparoscopic hernia repair procedures (De Novo DEN230084).
A coated stent to open the airway passage in the trachea (De Novo DEN230087).
A robotically assisted surgical device for endoscopic procedures (De Novo DEN230078).
An OTC blood pressure monitor with AFib detection feature (De Novo DEN230076).
A home-use rapid screening test for combined COVID-19 and Flu A/B. (De Novo DEN240029)
A catheter and ablation system for treatment of persistent atrial fibrillation. (PMA, Original, P240013).
A powered device to treat non-small cell lung cancer using tumor treatment fields (PMA, Original, P230042).
A stent system to reduce the risk of adverse events in carotid artery plaque removal procedures (PMA, Original, P240009).
An intraocular lens to mitigate the effect of presbyopia (PMA, Original, P240005).
A DNA test for colorectal cancer screening (PMA, Original, P230043).
A companion diagnostic assay for patients eligible to receive VYLOY therapy for treatment of gastric cancer (PMA, Original, P230018).
A total of 298 devices were cleared through the 510(k) pathway during October 2024. Days to FDA decision ranged from 7 to 1212 days with a median of 123 days. Top 5 medical specialties were Orthopedic (OR), Radiology (RA), General & Plastic Surgery (SU), Cardiovascular (CV), and Dental (DE) accounting for 62% (186/298) of devices cleared.
FDA warning letters
Rolence Ent. Inc.: Quality system violations - design controls, CAPA effectiveness, complaints handling, product acceptance status, software validation, labeling, device history records.
Molecular Testing Labs: Marketing without FDA authorization.
Class I recall announcements
Fresenius Kabi USA, LLC: software issues and cybersecurity vulnerability in large volume infusion system pump.
Philips Respironics: software issues in Trilogy ventilators.
Sentec/Percussionaire: use instructions update to avoid patient injuries due to use-errors in an IPV therapy device.
Mercury Medical: removal of certain emergency gas-powered resuscitators due to defects in the inline controller.
Zyno Medical: risk of air embolism in blood due to a software error in an infusion pump.
Medtronic: increased risk of reduced battery life in MiniMed 600 and 700 series insulin pumps.
Boston Scientific: increased risk of GI injury from the Obsidio conformable embolic agent.
Baxter Healthcare: increased risk of injury due to failure to issue low gas pressure alarm in a ventilator system.
Smiths Medical: manufacturing defect in a tracheostomy tube kit may cause pilot balloon to disconnect from the inflation line.
Kinova: increased risk of fire hazard due to broken insulation of a robotic assistive arm mounted on an electrically powered wheelchair.
GE HealthCare: risk of formaldehyde exposure from compressors used as ventilator accessories.
GE HealthCare:: risk of infant incubator and warmer door to become loose.
Let us take a closer look.
I. Innovation
Here is a summary of De-Novo decisions, PMA approvals and 510(k) clearances in the month of October 2024. See footnotes for links to additional information.
A. De-Novos
DEN23082: Stone Clear urinary stone propulsion device1
This ultrasonic propulsion device is used for the repositioning of residual stone fragments located in the upper urinary tract of adult patients to facilitate safe passage. Individual fragments must be less than or equal to 5 mm.Product Code QNA, Class II, Regulation 21 CFR 876.4690
DEN230084: Dexter L6 surgical system2
This electromechanical surgical system is intended to assist in accurate control of endoscopes and other instruments for tissue manipulation during hernia repair laparoscopic procedures.
Product Code SDD, Class II, Regulation 21 CFR 878.4965DEN230087: AMStent® Tracheobronchial Covered Stent System3
This device is a coated stent for use in the treatment of tracheobronchial narrowing due to growth of cancerous tumor.
Product Code SDB, Class II, Regulation 21 CFR 868.3721DEN230078: Versius Surgical System4
This device is a robotically assisted surgical device used for precise and accurate control of Versius Surgical endoscopic instruments during soft tissue minimal access surgery including cholecystectomy.
Product Code SCV, Class II, Regulation 21 CFR 878.4964DEN230076: Omron blood pressure monitor with AFib detection feature5
This device is a digital monitor intended for use in measuring blood pressure and pulse rate. It also detects any irregular heartbeats and provides a warning about episodes of atrial fibrillation (AFib) for the purpose of home-based monitoring and screening.
Product Code QXY, Class II, Regulation 21 CFR 870.1135DEN240029: Healgen Rapid Check COVID-19/flu A&B Antigen Test6
This device is a lateral flow immunoassay intended for qualitative detection and differentiation of COVID-19 and influenza A/B antigens in anterior nasal swab samples. It is intended for at-home use without a prescription.
Product Code SCA, Class II, Regulation 21 CFR 866.3987
B. Premarket Approvals (PMA) - Original only
1. P240013: Sphere-9™ Catheter and Affera™ Ablation System7
The Sphere-9 Catheter and Affera Ablation System are used in conjunction with the Affera Mapping System for cardiac mapping, ablation, and pacing. It is indicated for use in the treatment of recurrent atrial fibrillation which is non-responsive to medication and is symptomatic and persistent.
Figure 1 shows the key features of the Sphere-9 Catheter. An important feature is the spherical electrode lattice design with the ability to deliver both RF and PF energy for ablation.
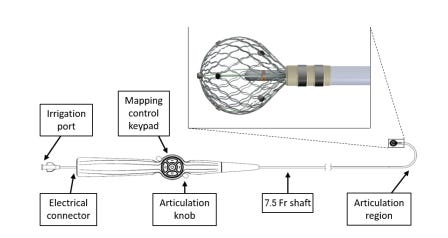
The Affera Ablation system includes the following components:
HexaGen RF Generator
HexaPulse PF Generator
HexaFlow Irrigation Pump
A prospective, multi-center, randomized clinical study (SPHERE Per-AF) treated 420 patients with a multi-visit follow-up schedule lasting a total of 360 days. Subjects were randomly assigned 1:1 to receive treatment with either the Sphere-9 Catheter with the Affera Mapping and Ablation System (investigational device) or the THERMOCOOL SMARTTOUCH® SF radiofrequency ablation catheter (control device).
The primary effectiveness endpoint was freedom from documented recurrence of atrial fibrillation (AF), atrial flutter (AFL), or atrial tachycardia (AT) based on electrocardiographic data through 12-month follow-up.
According to the Summary of Safety and Effectiveness Data (SSED)8:
Based on the SPHERE Per-AF clinical study, 74% of the investigational arm patients were free from all primary effectiveness failure events (including atrial arrhythmia recurrence) through 360 days after treatment with the Sphere-9 Catheter and Affera Ablation System. Further, mean change in quality of life from baseline to 360 days post ablation was consistent with a clinically significant improvement.
The incidence of primary adverse events was 1.4% requiring hospitalization.
FDA approved the PMA on October 24, 2024 with a requirement for a post-approval study.
2. P230042: Optune Lua™ tumor treatment fields generator9
Optune Lua(TM) is a portable, powered device which produces alternating electrical fields to physically disrupt the rapid cell division of cancerous cells. It is indicated in patients with metastatic non-small cell lung cancer in combination with PD-1/PD-L1 inhibitors or docetaxel.

A pivotal clinical study (LUNAR) enrolled 534 patients with stage 4 advanced/metastatic non-small cell lung cancer (NSLC). It was a prospective, randomized, open-label multi-center study, with 1:1 randomization with the investigative device and standard of care ((TTFields+SOC) vs. standard of care alone (SOC).
The primary effectiveness endpoint was median overall survival (OS) of patients in the treatment arm compared to patients in the control arm. According to the Summary of Safety and Effectiveness Data(SSED)10:
At the final analysis in the ITT population, median OS in the TTFields+SOC group was 13.2 months (95% CI, 10.3 to 15.5) compared to 9.9 months (95% CI, 8.2 to 12.2) in the SOC group. The hazard ratio of death was 0.76 (95% CI, 0.58 to 0.99; P=0.042). The 1-year OS rate was 53% (95% CI, 44 to 61) with TTFields+SOC, and 42% (95% CI, 34 to 50) with SOC. The study met its pre-specified threshold for statistical significance (P=0.041) at the final analysis.
Further, the LUNAR study demonstrated that TTFields therapy was well-tolerated, and did not result in adverse interactions with SOC therapies.
FDA approved the PMA on October 15, 2024.
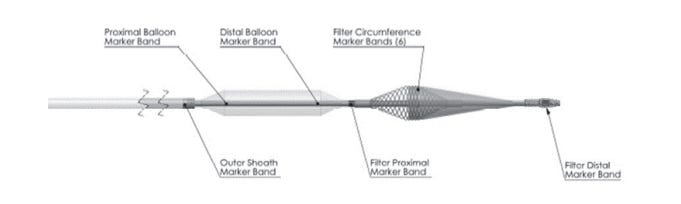
3. P240009: Neuroguard IEP® System11
The Neuroguard IEP system consists of 3 main components:
A self-expanding nickel-titanium stent
A delivery catheter with a post-dilation balloon and activation handle
Embolic protection filter
This system is indicated for use in patients who meet certain criteria and are at high risk of adverse events from carotid endarterectomy (CEA), a surgical procedure to remove plaque buildup in the carotid artery to improve blood flow to the brain.

A prospective, multi-center, single-arm clinical study enrolled 305 patients at 23 US sites and 9 OUS sites. According to the Summary of Safety and Effectiveness Data (SSED)12:
Effectiveness of the device was analyzed by evaluating the composite primary endpoint of major adverse events (MAE) consisting of death, stroke and myocardial infarction through 30 days and one-year ipsilateral stroke. Given the weighted performance goal of 13.8% and expected primary endpoint event rate of 8.1%, the observed MAE rate of 2.84 % in the intention-to-treat population met the performance goal (p<0.05, 1-sided test).
FDA approved the PMA on October 11, 2024 with a requirements for a post-approval study.
4. P240005: enVista Envy™ intraocular lens (IOL)13
This intraocular lens (IOL) is indicated for primary implantation in the capsular bag of the eye for visual correction of aphakia with less than or equal to 1.0 Diopter pre-operative corneal astigmatism following removal of a cataractous lens to mitigate the effect of presbyopia.
A multi-center, randomized, and controlled clinical trial with a 1-year follow-up demonstrated excellent long term outcomes14. It also showed superior tolerance to glares, halos and starbursts compared to PanOptix.

FDA approved the PMA on October 10, 2024. The Summary of Safety and Effectiveness Data (SSED) is not publicly available yet.
5. P230043: Cologuard Plus™ Colorectal Cancer Screening Test15
The Cologuard Plus test is an in vitro diagnostic device designed to analyze a patient’s stool for the presence of DNA and hemoglobin markers which may indicate the presence of colorectal cancer (CRC) or advanced precancerous lesions (APL).
The Cologuard Plus system consists of a collection kit, reagents and various instruments qualified by Exact Sciences under the Exact Quality System.
A prospective, cross-sectional, multi-center primary clinical study was conducted with 26,758 subjects across 186 sites in the US. According to the Summary of Safety and Effectiveness Data (SSED)16:
The study conducted demonstrated probable benefit for CRC detection and detection of advanced precancerous lesions (APL). The sensitivity for CRC was observed to be 95.3% (81/85, 2-sided 95% CI: 88.4-98.7%) and the sensitivity for APL was 43.3% (849/1962, 2-sided 95% CI: 41.1-45.5%). The specificity was 90.7% (849/1962, 95% CI: 90.3%-91.1%).
FDA approved the PMA on October 3, 2024.
6. P230018:VENTANA CLDN18 (43-14A) RxDx Assay17
This device is a qualitative assay used with OptiView DAB IHC detection kit indicated as an aid in identifying patients with gastric or GEJ adenocarcinoma who may be eligible for treatment with VYLOY (zolbetuximab) in accordance with the approved therapeutic product labeling.
FDA approved the PMA on October 18, 2024. The Summary of Safety and Effectiveness Data (SSED) is not publicly available yet.
C. Premarket Notifications (510k)
FDA cleared a total of 298 devices through the premarket notification process during October 2024. A majority of these devices (78%) were cleared through the Traditional 510k process.
Following graphic provides an overview of these 510k’s.

Here are the key highlights:
Top 5 medical specialties accounted for 62% of the total 510k (186/298) with median days to decision ranging from 55 to 178 days after receipt.
Orthopedic (OR) medical specialty had the highest number of 510k (47/298) with 55 median days to decision after receipt. Top 5 product codes were in the Orthopedic, Radiology, General & Plastic Surgery and Dental medical specialties.
QIH was the top product code corresponding to a an automated radiological image processing software in the Radiology category with 8 510k clearances. Product code MAX corresponding to intervertebral fusion device in the Orthopedic specialty also had 8 510k clearances.
In aggregate, days to decision ranged from a minimum of 7 days to a maximum of 1212 days after receipt, with a median of 123 days. 20th percentile of the devices were cleared within 42 days, while the 80th percentile were cleared within 239 days.
IceSeed, IceSphere and IceRod cryosurgical needles for general surgery were cleared just 7 days after receipt (K243245, PRO Code: GEH).
It took 1212 days for a Microlyte Ag/Lidocaine wound dressing to receive 510k clearance as a Class II device (K211943, PRO Code: FRO).
II. FDA Warning Letters
FDA issued 2 warning letters to medical device manufacturers during the month of October 2024.
CMS 695010 - Rolence Ent. Inc.18
Devices impacted: XR-01 portable x-ray system.
Observations cited:
Quality system violations - design controls, CAPA effectiveness, complaints handling, product acceptance status, software validation, labeling, device history records
Adverse events reporting violations.
CMS 680324 - Molecular Testing Labs19
Devices impacted: HIV serological diagnostic dried blood spot card self-collection kit.
Observations cited:
Marketing without FDA authorization
III. Medical Device Recalls
In the month of October 2024, FDA published 12 Class I recalls. This classification reflects the most serious type of recall, where the use of impacted devices may cause serious injuries or death.
Fresenius Kabi USA, LLC: Ivenix Infusion System Large Volume Pump (LVP) Software20
Device use: the Ivenix infusion system is used for controlled delivery of fluids in a hospital or outpatient care setting.
Reasons for recall: software correction needed to address multiple software issues and a cybersecurity vulnerability.
Patient safety impact: delay or underdosage of therapy may occur due to these issues. Use of affected devices may cause serious health effects including arrhythmia, hyperglycemia, low or high blood pressure, undersedation, blood clotting changes and death. There has been no reports of injury or death.
Philips Respironics: Trilogy ventilators21
Device use: these devices are used to provide breathing support in a continuous or intermittent mode. They are used in medical, home, and non-emergency transport settings.
Reasons for recall: software correction needed to address multiple software issues related to safety.
Patient safety impact: use of affected devices may cause serious health issues including hypoventilation, hypoxia and death. There have 9 reported injuries and 1 death related to this issue.
Sentec/Percussionaire: Phasitron 5 In-Line Valve22
Device use: this in-line valve is a component of the Phasitron 5 system used to provide Intrapulmonary Percussive Ventilation (IPV) therapy to patients on mechanical ventilation.
Reasons for recall: IFU update following increase in reports of patient injuries due to use-errors.
Patient safety impact: use of affected devices without following updated instructions may cause serious health issues, including cardiac arrest, pneumothorax, tracheobronchial tear and death. There have 6 reported injuries but no reported deaths.
Mercury Medical: Neo-Tee T-Piece Resuscitators23
Device use: these devices are gas-powered resuscitators used in an emergency to provide breathing support through a face mask or a tube. It is also used with newborns and infants.
Reasons for recall: potential for the inline controller to come apart causing a loss of positive pressure.
Patient safety impact: use of affected devices may cause serious health issues including ischemia and death. There has been no reported injuries or death related to this issue.
Zyno Medical: Infusion Pumps24
Device use: these infusion systems are used to deliver nutrition or essential fluids, blood and related products.
Reasons for recall: software error in the air-in-line software may cause air bubbles to be introduced in the blood stream.
Patient safety impact: use of affected products may serious health issues including vascular air embolism, fast and irregular heartbeat, heart attack, stroke, seizure and death. There is a report of 2 injuries but no deaths related to this issue.
Medtronic: MiniMed 600 and 700 Series Pumps25
Device use: these battery-operated insulin pumps are used for management of diabetes in patients on insulin therapy.
Reasons for recall: increased risk of reduced battery life.
Patient safety impact: use of affected devices may cause serious health issues including hyperglycemia, diabetic ketoacidosis and death. There have been 170 reports of hyperglycemia, 11 reports of diabetic ketoacidosis but no reported deaths due to this issue.
Boston Scientific: Obsidio Conformable Embolic26
Device use: this device is a pre-mix embolic agent used to block blood flow to specific blood vessels to stop bleeding or hemorrhaging.
Reasons for recall: new risk of adverse events in the total GI area.
Patient safety impact: use of affected devices may cause serious health issues including off-target embolization, ischemia, and prolonged hospitalization. There have been 15 reports of injury and 4 deaths related to this issue.
Baxter Healthcare: Life2000 Ventilation System27
Device use: this device is used to provide continuous or intermittent breathing support.
Reasons for recall: update of use instructions due to the risk of failing to issue a low gas pressure alarm.
Patient safety impact: use of affected devices may cause serious health issues including shortness of breath, desaturation, and death. There have been 1 reported injury but no deaths related to this issue.
Smiths Medical: Tracheostomy Tube Kits28
Device use: this device is used to provide an artificial airway after trauma or due to a medical condition.
Reasons for recall: a manufacturing defect may cause the pilot balloon to disconnect from the tracheostomy inflation line.
Patient safety impact: use of affected devices may cause serious health issues including aspiration and death. There have been 12 reported injury but no deaths related to this issue.
Kinova: Jaco Assistive Robotic Arm29
Device use: this robotic arm device is used to support patients who have lost most or all functionality of their arms. It is designed to be installed on a motorized wheelchair, and is controlled through the wheelchair’s drive controls.
Reasons for recall: increased risk of fire hazard due to contact with electric circuitry.
Patient safety impact: use of affected devices may cause serious health issues including burns, other thermal injuries and death. There have been no reported injury or deaths related to this issue.
GE HealthCare: EVair Compressors30
Device use: this device is an optional accessory to ventilators used as an alternate source of air flow for patients. It is intended to be connected to a Datex-Ohmeda Inc. critical care ventilator as a supply of compressed medical breathing air.
Reasons for recall: new use instructions to manage risk of formaldehyde exposure.
Patient safety impact: use of affected devices may cause serious health issues including airway irritation or inflammation with higher risk to newborns and infants. There have been no reported injury or deaths related to this issue.
GE HealthCare: Giraffe OmniBed and Giraffe Omnibed CareStation31
Device use: this device are used as combination infant incubator and infant warmer.
Reasons for recall: risk of loose heater doors due to improper screw closure.
Patient safety impact: use of affected devices may cause serious health issues and minor trauma in the neonatal patient, including deep soft tissue injuries, traumatic brain injury and death. There have been no reported injury or deaths related to this issue.
FDA: DEN230082 De Novo database entry.
FDA: DEN230084 De Novo database entry.
FDA: DEN230087 De Novo database entry.
FDA: DEN230078 De Novo database entry.
FDA: DEN230076 De Novo database entry.
FDA: DEN240029 De Novo database entry.
FDA: P240013 Sphere-9™ Catheter and Affera™ Ablation System PMA database entry.
FDA: P240013 Summary of Safety and Effectiveness Data.
FDA: P230042 Optune Lua™ PMA database entry.
FDA: P230042 Summary of Safety and Effectiveness Data.
FDA: P240009 Neuroguard IEP® System PMA database entry.
FDA: P240009 Summary of Safety and Effectiveness Data.
FDA: P240005 enVista Envy™ intraocular lens (IOL) PMA database entry.
Bausch & Lomb: enVista ENVY webpage, accessed on 21 November 2024.
FDA: P230043 Cologuard Plus™ PMA database entry.
FDA: P230043 Summary of Safety and Effectiveness Data (SSED).
FDA: P230018 VENTANA CLDN18 (43-14A) RxDx Assay PMA database entry.
FDA: Rolence Ent. Inc. warning letter.
FDA: Molecular Testing Labs warning letter.
FDA: Fresenius Kabi USA, LLC recall announcement.
FDA: Philips Respironics recall announcement.
FDA: Sentec/Percussionaire recall announcement.
FDA: Mercury Medical recall announcement.
FDA: Zyno Medical recall announcement.
FDA: Medtronic recall announcement.
FDA: Boston Scientific recall announcement.
FDA: Baxter Healthcare recall announcement.
FDA: Smiths Medical recall announcement.
FDA: Kinova recall announcement.
FDA: GE Healthcare recall announcement.
FDA: GE Healthcare recall announcement.