



Medical Device News Update - August 2024
FDA approvals, warning letters and Class I recalls issued during August 2024

Monthly roundup of FDA approvals, warning letters and recalls issued in August 2024

Each month, Let’s Talk Risk! (LTR) provides a comprehensive summary of new device approvals or clearances, warning letters, and Class I recall announcements issued by the FDA in the previous month.
As a risk practitioner, it is very important to stay up to date with the latest regulatory developments affecting the medical device industry. A review of latest innovations approved or cleared by the FDA helps in understanding different ways the agency evaluates benefit-risk of medical devices when reviewing their safety and effectiveness. Warning letters and recall announcements are useful in building awareness of gaps in quality management system and the risk management process that should be addressed before they are cited by the FDA.
Only publicly available information is curated in this article. Links to relevant sources are provided in the footnotes.
Quick Summary
New device approvals/clearances
A high throughput genetic sequencing based tumor profiling test (De Novo DEN230046).
A device to reduce bacterial load on respiratory devices and accessories (De Novo DEN210037).
A home-based test for qualitative detection of syphilis (De Novo DEN230090).
A rapid UV disinfection chamber for ultrasound probes (De Novo DEN230067).
An implantable nerve stimulation device to reduce post-amputation pain, breakthrough device designation (PMA, Original, P230020).
A stent system for treatment of pulmonary artery stenosis, breakthrough device designation (PMA, Original, P240003).
A companion diagnostic assay for qualitative detection of 517 genetic variants in tumor tissues from patients considered for VITRAKVI® and RETEVMO® therapies (PMA, Original, P230011).
A companion diagnostic assay for detection of MAGE-A4 protein as an aid in identifying patients considered for TECELRA® immunotherapy (PMA, Original, P230016).
A total of 283 devices were cleared through the 510(k) pathway during August 2024. Days to FDA decision ranged from 1 to 1062 days with a median of 116 days. Top 5 medical specialties were Orthopedic (OR), Radiology (RA), General & Plastic Surgery (SU), Cardiovascular (CV), and Dental (DE) accounting for 62% (176/283) of devices cleared.
FDA warning letters
Shenzhen Moyeah Intelligent Life Technology Co.: Marketing of various CPAP cleaning products and accessories without FDA authorization.
Adventure Innovations LLC: Marketing of UV based cleaning/disinfecting devices for CPAP and accessories without FDA authorization.
Natures Pillows, Inc. and Top Dog Direct, LLC: Marketing of sanitizers for CPAP and accessories without FDA authorization.
LEEL Tech: Marketing of sanitizers for CPAP and accessories without FDA authorization.
Class I recall announcements
Smiths Medical: multiple issues with older versions of the software in the CADD-Solis and CADD-Solis VIP Ambulatory infusion pumps.
Medtronic: correction for NIM Vital Nerve Monitoring System due to the potential for false negative response.
Abiomed: removal of 9 Impella CP with SmartAssist Systems due to release of products that failed QC inspection.
ICU Medical: diminished battery life due to manufacturing error which can impact infusion pump operation.
Defibtech, LLC: motor failure in a chest compression device.
Medline Industries, LP: defective syringes in convenience kits that may break or leak.
Inari Medical: patient injuries and deaths due to entrapment of a clot removing catheter.
Let us take a closer look.
I. Innovation
Here is a summary of De-Novo decisions, PMA approvals and 510(k) clearances in the month of August 2024. See footnotes for links to additional information.
A. De-Novos
DEN230046: PGDx elio plasma focus Dx1
A high throughput genetic sequencing based tumor profiling test for detection of circulating cell-free nucleic acid fragments for monitoring of cancer.Product Code SBY, Class II, Regulation 21 CFR 866.6085
DEN210037: SoClean 3+ Bacterial Reduction Device2
This device is used to clean home-use CPAP masks and accessories to reduce the bacterial load.
Product Code QXQ, Class II, Regulation 21 CFR 880.6993DEN230090: First To Know Syphilis Test3
This is a test for qualitative detection of antibodies for syphilis for use in home settings as a screening tool for further diagnosis and treatment by a healthcare professional.
Product Code SBZ, Class II, Regulation 21 CFR 866.3986DEN230067: Chronos® UV radiation chamber disinfection device4
This device uses UV for rapid disinfection of ultrasound probes used in various medical/surgical procedures.
Product Code OSZ, Class II, Regulation 21 CFR 880.6511
B. Premarket Approvals (PMA) - Original only
1. P230020: Altius® Direct Electrical Nerve Stimulation System5
This direct electrical nerve stimulation device received the FDA breakthrough device designation on April 30, 2021. It is used for management of chronic intractable phantom and residual lower limb pain in adult amputees.
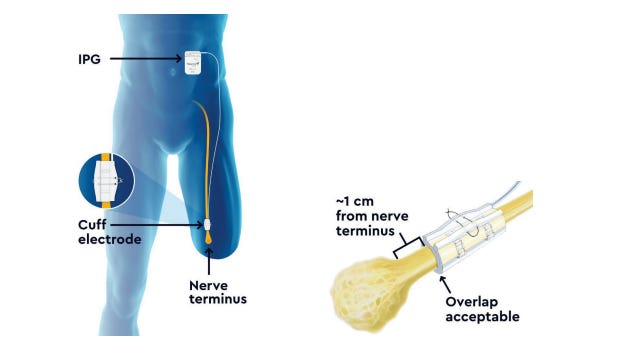
The Altius system includes the following components:
Implantable pulse generator (IPG) and Nerve cuff electrode
Patient controller and battery charger
Clinician programmer wand and application
Torque wrench
A prospective, multicenter, randomized, sham-controlled, double blind QUEST study with 180 subjects was conducted for evaluation of safety and effectiveness.
According to the Summary of Safety and Effectiveness Data (SSED)6, a systematic review of clinical literature was used to establish reasonable assurance of safety and effectiveness:
The QUEST study met its pre-specified primary effectiveness endpoint, demonstrating superior pain relief with the active Altius treatment (Test) compared to sham control, and the study was deemed a success with respect to effectiveness. The absolute difference between the treatment arms in terms of responder rate at Month 3 (i.e., treatment effect) was 17.6% in favor of active Altius treatment, and this difference was highly statistically significant. The benefits observed during the blinded Randomized Testing phase continued to increase through one year
FDA approved the PMA on August 26, 2024.
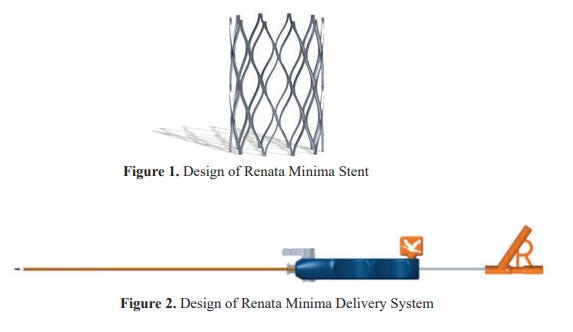
2. P240003: Minima Stent System7
The Minima Stent system received the breakthrough device designation from the FDA on February 7, 2022. It is indicated for use in the treatment of native or acquired pulmonary artery stenoses or coarctation of the aorta in neonates, infants, and children at least 1.5 kg in weight.
The Minima stent system includes the following components:
The Minima stent
The Minima delivery system: balloon catheter, outer covering catheter
According to the Summary of Safety and Effectiveness Data (SSED)8:
As evidenced by the results of the GROWTH study, the performance goal of the primary endpoint was met, with 41 out of the 42 subjects (97.2%) meeting the criteria for clinical success at the 6-month follow-up visit.
Further:
The results from the non-clinical laboratory and animal testing demonstrate that the device is suitable for long-term implant. The safety assessment for the GROWTH Trial was based on freedom from procedure- or device-related serious adverse events.
Based on these results, FDA approved the PMA on August 28, 2024, with continued follow-up and a new post-approval study.
3. P230011: TruSight™ Oncology Comprehensive9
This device was granted the breakthrough device designation by the FDA on January 17, 2019. It is a qualitative in-vitro diagnostic test that uses next generation sequencing (NGS) to detect variants of 517 genes in tumor tissue samples from cancer patients with solid malignant neoplasms. It is intended to be used as a companion diagnostics to identify patients who may benefit from VITRAKVI® and RETEVMO® therapies.
It is validated for use on the Illumina NextSeq 550Dx instrument.
4. P230016: MAGE-A4 IHC 1F9 pharmDx10
This is a qualitative assay intended for use in detection of MAGE-A4 protein as an aid in identifying adult patients with synovial sarcoma for whom TECELRA, a T-cell immunotherapy is being considered.
C. Premarket Notifications (510k)
FDA cleared a total of 283 devices through the premarket notification process during August 2024. A majority of these devices (80%) were cleared through the Traditional 510k process.
Following graphic provides an overview of these 510k’s.
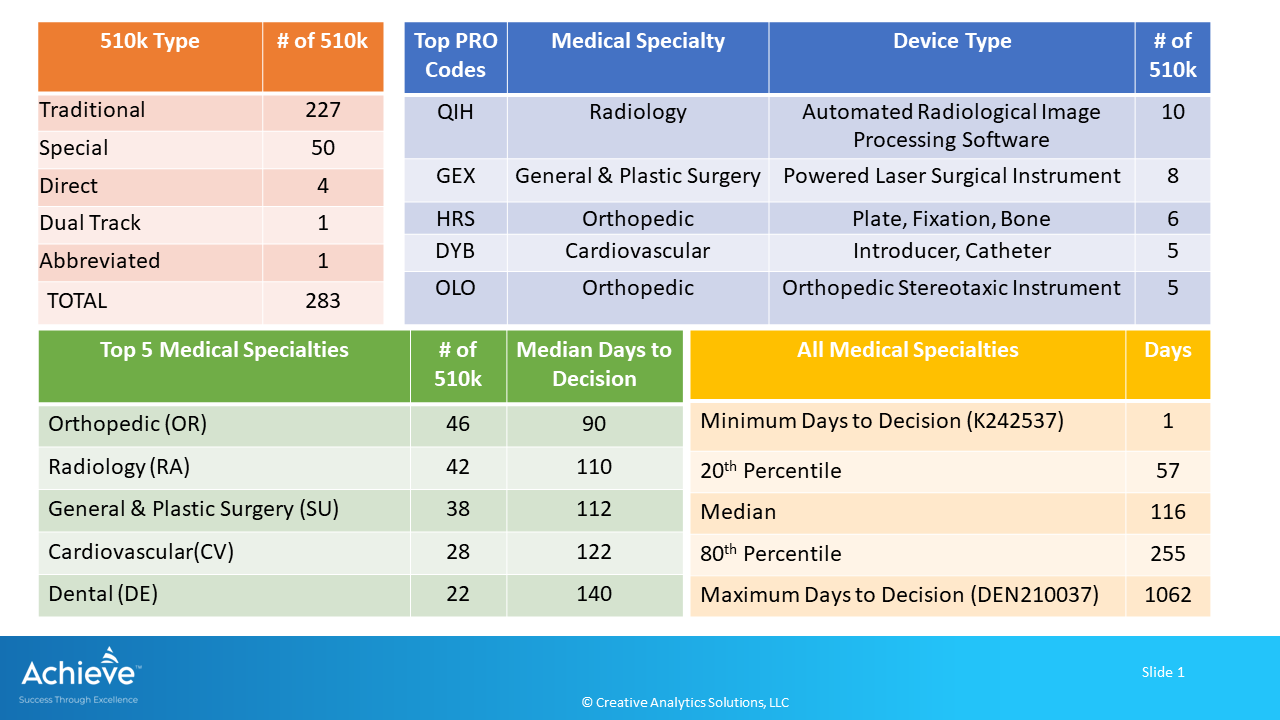
Here are the key highlights:
Top 5 medical specialties accounted for 62% of the total 510k (176/283) with median days to decision ranging from 90 to 140 days after receipt.
Orthopedic (OR) medical specialty had the highest number of 510k (46/283) with 90 median days to decision after receipt. Top 5 product codes were in the Radiology, General & Plastic Surgery, Orthopedic and Cardiovascular medical specialties.
QIH was the top product code corresponding to an automated radiological image processing software in the Radiology category with 10 510ks.
In aggregate, days to decision ranged from a minimum of 1 day to a maximum of 1062 days after receipt, with a median of 116 days. 20th percentile of the devices were cleared within 57 days, while the 80th percentile were cleared within 255 days.
A brace fluoride sealant was cleared just a day after receipt (K242537, PRO Code: DYH).
It took 1062 days for a bacterial reduction device to receive De Novo classification as a Class II device (DEN210037, PRO Code: QXQ).
II. FDA Warning Letters
FDA issued 4 warning letters to medical device manufacturers during the month of August 2024.
CMS 677092, Shenzhen Moyeah Intelligent Life Technology Co.11
Devices impacted: Various cleaning accessories for CPAP devices.
Observations cited:
Marketing without FDA authorization.
CMS 676842, Adventure Innovations LLC12
Devices impacted: Sani Bot D3 UV cleaning/disinfecting device for CPAP devices and accessories
Observations cited:
Marketing without FDA authorization.
CMS 676849, Natures Pillows, Inc. and Top Dog Direct, LLC13
Devices impacted: Clean Zone CPAP Sanitizer
Observations cited:
Marketing without FDA authorization.
CMS 677297, LEEL Tech14
Devices impacted: CLYN (CZ001& CLYN Universal), LEEL, and SOLID (Solid CPAP & ONECLICK SOLID) for CPAP devices and accessories
Observations cited:
Marketing without FDA authorization.
III. Medical Device Recalls
In the month of August 2024, FDA published 7 Class I recalls. This classification reflects the most serious type of recall, where the use of impacted devices may cause serious injuries or death.
Smiths Medical: Ambulatory Infusion Pump Software Correction15
Device use: the CADD Solis and CADD Solis VIP Ambulatory infusion pumps are used for delivery of medication at continuous infusion rates, intermittent bolus or patient controlled doses.
Reasons for recall: software errors in versions before v4.3 leading to various issues in pump operation and alarms.
Patient safety impact: patients may experience serious adverse health consequences related to delay, interruption, under- or over-administration of therapy, or death. There has been 1 reported injury but no deaths.
Medtronic: Nerve Monitoring System16
Device use: NIM vital nerve monitoring system is used to locate, monitor and stimulate nerves in the skull and spine during surgery.
Reasons for recall: reports of false negative response where the device fails to issue an electromyography (EMG) tone when the NIM probe is placed on a nerve.
Patient safety impact: this issue may cause serious adverse health consequences, including nerve damage, facial nerve damage, nerve weakening (paresis), and nerve paralysis. There have 10 reported injuries but no deaths related to this issue.
Abiomed: Impella CP with SmartAssist Systems17
Device use: these pumps are used for short term to provide ventricular support for adequate blood circulation during high-risk procedures.
Reasons for recall: 9 pumps failed quality control inspection but released to customers.
Patient safety impact: use of affected pumps can cause serious life threatening injuries including death. There have no reported injuries or death and none of the recalled devices were used in patients.
ICU Medical: Batteries in Plum 360, A+ and A+3 Infusion System18
Device use: these are large volume infusion pumps used to deliver fluids, medications and blood products to patients at controlled rates.
Reasons for recall: manufacturing defect in batteries which can result in diminished battery life.
Patient safety impact: use of affected products can cause serious injury due to pump operational issues. There has been 1 reported injuries but no reports of death related to this issue.
Defibtech, LLC: RMU-2000 ARM XR Chest Compression Device19
Device use: this device is used to provide chest compression in adults whose hearts suddenly stop functioning.
Reasons for recall: problem with the device motor that may cause it to fail.
Patient safety impact: failure to revive a patient’s heart may lead to serious life threatening injury including death. There is a report of 1 injury but no deaths related to this issue.
Medline Industries, LP: Convenience Kit Syringes Manufactured in China20
Device use: convenience kits containing syringes used in various surgical procedures.
Reasons for recall: syringes may break or leak.
Patient safety impact: use of potentially defective syringes can cause infection or blood vessel blockage, and other life threatening injuries including death. There have been no reports of injury or death due to this issue.
Inari Medical: ClotTriever XL Catheter21
Device use: ClotTriever XL Catheter is used to remove clots and blockages from large blood vessels outside the heart, including the vena cava.
Reasons for recall: injuries caused by device entrapment leading to blockage of lung arteries.
Patient safety impact: serious and life threatening injuries including death may occur. There have been 4 reports of injuries and 6 deaths due to this issue.
FDA: DEN230046 De Novo database entry.
FDA: DEN210037 De Novo database entry.
FDA: DEN230090 De Novo database entry.
FDA: DEN230067 De Novo database entry.
FDA: P230020 Altius® Direct Electrical Nerve Stimulation System PMA database entry.
FDA: P230020 Summary of Safety and Effectiveness Data.
FDA: P240003 Minima Stent System PMA database entry.
FDA: P240003 Summary of Safety and Effectiveness Data.
FDA: P230011 TruSight™ Oncology Comprehensive PMA database entry.
FDA: P230016 MAGE-A4 IHC 1F9 pharmDx PMA database entry.
FDA: Shenzhen Moyeah Intelligent Life Technology Co. Warning Letter.
FDA: Adventure Innovations LLC Warning Letter.
FDA: Natures Pillows, Inc. and Top Dog Direct, LLC, Warning Letter.
FDA: LEEL Tech, Warning Letter.
FDA: Smiths Medical recall announcement.
FDA: Medtronic recall announcement.
FDA: Abiomed recall announcement.
FDA: ICU Medical recall announcement.
FDA: Defibtech, LLC recall announcement.
FDA: Medline Industries, LP recall announcement.
FDA: Inari Medical recall announcement.