



QMSR Tip #4: Review and fix these disconnects between risk management and design controls
Recent FDA inspection data and warning letters highlight a significant disconnect between risk management and design controls. Review and fix these disconnects as you get ready for the QMSR.


FDA requires manufacturers to establish and maintain procedures to control the design of their medical devices in order to ensure that specified design requirements are met. These design controls requirements are specified in 21 CFR 820.30 (a) through (j)1.
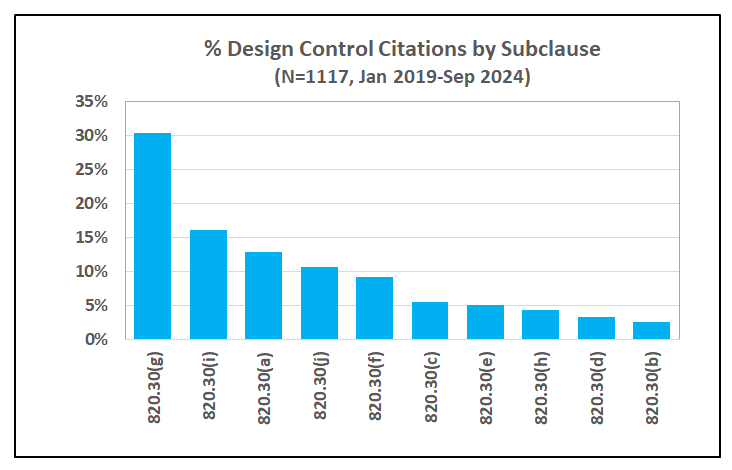
Design Controls also happens to be one of the top categories of inspectional observations, according to recent FDA data2, accounting for an average 13% of the total observations cited in the last 5 years. As shown in Figure 1 below, FDA issued a total of 1117 observations to medical device manufacturers during inspections ending between January 2019 through September 20243. On a percentage basis, deficiencies in design validation, design changes, general controls, design history file and design verification contributed to nearly 80% of these inspectional observations.
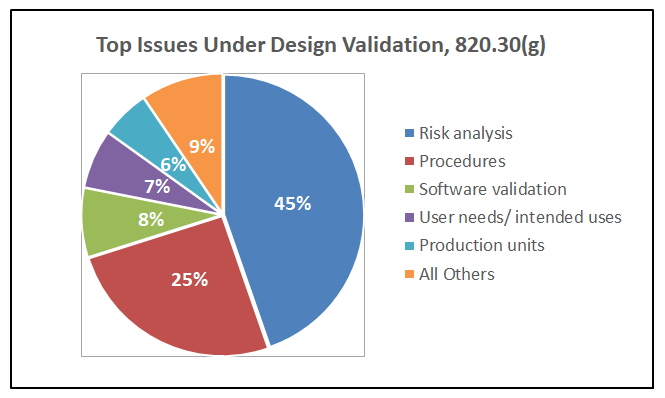
A closer look at the top issues in design validation, 21 CFR 820.30(g), reveals that deficiencies in risk analysis account for 45% of the 338 observations in this category.
As you prepare your transition to the Quality Management System Regulation (QMSR), it is important to appreciate that FDA scrutiny on risk management will only increase over time. In the current Quality System (QS) regulation, risk is covered only in one sub-clause, that is in 21 CFR 820.30 (g) for design validation. The QMSR however, is based on ISO 13485:2016, which has many more specific requirements for risk management across the entire quality system.
This article highlights 5 potential disconnects between risk management and design controls. Although not a comprehensive list, we think these are the most important areas of opportunity based on our analysis of recent FDA warning letters. We recommend that you review your current Quality Management System (QMS) for these disconnects and identify actions to remedy them as part of your transition plan to the QMSR.
Let us review the design control requirements according to the current Quality System (QS) regulation
FDA has specified the current requirements for design controls in 21 CFR 820.30. Intended to cover the design and development activities throughout the lifecycle of a medical device, these requirements are further classified in 10 sub-clauses as summarized in the Figure below.

It is interesting to note that there is only one requirement for risk analysis in the entire design control clause under 21 CFR 820.30(g) applicable to design validation:
Design validation shall include software validation and risk analysis, where appropriate.
In fact, there is no other risk-related requirement explicitly stated in the entire QS regulation! However, FDA offers a considerable amount of commentary in the preamble4 to the QS regulation to clarify their approach to risk management and expectations from device manufacturers. As an example, FDA writes in response to comment 83 in the preamble:
Risk analysis must be conducted for the majority of devices subject to design controls and is considered to be an essential requirement for medical devices under this regulation, as well as under ISO/CD 13485 and EN46001.
It would appear that the practice of risk management, as a general observation across the medical device industry, has not been up to par with FDA’s expectations. FDA inspectional observations, as noted above, have routinely pointed to numerous deficiencies that are indicative of systemic gaps in the practice of risk management as a discipline.
Part of it could be because of the way the current QS regulation is written, which limits the Agency’s enforcement authority to a very narrow scope of risk analysis only under design controls. Another reason could be that combining risk analysis with software validation may give an impression that this requirement applies only to software and not all devices.
Risk management is more than risk analysis; it applies to all aspects of the QMS, not just design and development, as reflected by the current state-of-the-art International Standards such as ISO 134855 and ISO 149716. By incorporating ISO13485:2016 in the QMSR as a normative reference, FDA will have regulatory authority to enforce a risk-based approach throughout the quality system.
ISO 13485:2016 emphasizes risk across the entire QMS, including design and development
As a foundational principle, ISO 13485:2016 requires a risk-based approach per clause 4.1.2 (b):
4.1.2 The organization shall:
(b) apply a risk-based approach to the control of the appropriate processes needed for the quality management system.
Clearly, design and development is a critical process in the quality system, expected to deliver safe and effective devices that also comply with applicable regulatory requirements. As a result, there are additional specific risk-based requirements for design and development (clause 7.3), which is part of the product realization process covered in clause 7 of ISO 13485:
The organization shall document one or more processes for risk management in product realization. Records of risk management activities shall be maintained (see 4.2.5).
Additionally, in clause 7.3.3 for design and development inputs:
Inputs relating to product requirements shall be determined and records maintained (see 4.2.5). These inputs shall include:
c) applicable output(s) of risk management;
And in clause 7.3.9 for control of design and development changes
The review of design and development changes shall include evaluation of the effect of the changes on constituent parts and product in process or already delivered, inputs or outputs of risk management and product realization processes.
Further, it should be understood that where planning is required, for example design and development planning, a risk-based approach is implicitly expected.
ISO 13485, therefore, expects a tight integration of risk management activities with design and development. As a result, it is no longer sufficient to focus only on risk analysis as required by the current QS regulation. This requires a significant shift in the mindset, and a more thoughtful, systematic approach to building a solid competence in risk management.
Let us now review 5 potential disconnects between risk management and design controls with this perspective
As shown in Figure 1 above, design validation is the top category of inspectional observations, accounting for 30% of the 1117 observations issued by the FDA from 2019 through September 2024. Further, deficiencies in risk analysis, characterized by the FDA as risk analysis not performed or inadequate, accounted for 45% of the findings in design validation.
Readers of this Let’s Talk Risk! newsletter know that we compile a comprehensive summary of FDA approvals, warning letters and recalls each month to share the most recent MedTech news. Below are 5 key disconnects between risk management and design activities we have noticed in recent warning letters.
Disconnect #1: Missing hazards from risk assessments
In a warning letter issued to a manufacturer of cardiac heater-cooler product7, FDA cites the following observations
Your firm has not adequately conducted risk analysis for your MCH devices, as required by 21 CFR 820.30(g). Specifically, your firm added a new hazard of “Bacteria such as m. chimaera or other biological agents being aerosolized into patient environments” to your MCH-10ARH Risk Assessment document on August 6, 2021; however, your firm has been aware of this hazard since at least 2018. Additionally, this hazard has not been considered as part of your design activities.
Ironically, FDA issued this warning letter in the context of certain design changes that were intended to reduce the risk of infection by adding an optional airflow hood.
Specifically, your firm has made significant changes that include, among other things, the addition of an optional airflow hood, a dripless external hose kit, and thermoelectric cooling technology in certain MCH models. The addition of an optional airflow hood is intended to reduce the risk of infection via aerosolization of contaminated water. The airflow hood impacts how potentially contaminated water droplets are dispersed in the operating room.
If all known and potential hazards are not explicitly included in risk assessments, there is a chance that adequate risk controls are not considered, and testing not performed to ensure they are effective. In this case, this disconnect also led to inadequate design validation of changes intended to control the risk of bacterial infection.
Key point: ISO 14971 requires traceability of each identified hazard to risk analysis, risk evaluation, implementation and verification of risk control measures, and the results of the evaluation of the overall residual risk8.
A direct linkage between the outputs of risk analysis, specifically hazard analysis, and design inputs is needed. Think of this as a map of hazards, hazardous situation and harms and how they are linked to different design inputs. An industry best practice is to develop an efficient system of cross-referencing each risk item in a risk trace matrix with one or more design inputs/outputs in a design input trace matrix.
Disconnect #2: Failing to update risk analysis following M&A’s
In a warning letter issued to a manufacturer of a patient vital signs monitor9, FDA cites the following observation:
Your firm has not adequately conducted a risk analysis for your nGenuity devices, as required by 21 CFR 820.30(g). Specifically, your firm provided a Hazard Analysis & Management document for your nGenuity CO2 Project, dated 2007, which was a document from the company you purchased in 2015. This did not identify risks associated with distorted displays or devices shutting down unexpectedly. Since 2016, your firm received 629 complaints related to distorted displays and at least 21 complaints related to monitors shutting down unexpectedly with your nGenuity device.
Also, the above 2007 Hazard Analysis & Management document for your nGenuity CO2 Project, did not identify any hazards related to faulty internal ECG cables, which were incorporated into certain nGenuity devices after 2015.
In the same warning letter, FDA also cites additional observations related to inadequate design verification and validation. These observations collectively reflect a serious concern with the lack of control over the design of these devices.
Key point: Mergers and Acquisitions (M&A) are common in the medical device industry. After acquiring another company, the device manufacturer is responsible for continued safety and effectiveness of the acquired devices. This includes actively updating all relevant documentation, especially those related to risk management and design controls.
Disconnect #3: Inadequate analysis of complaints data for analyzing risk
In a warning letter issued to a manufacturer of orthopedic implants and accessories10, FDA cites the following observation:
Failure to establish and maintain design validation procedures to ensure proper risk analysis is completed, as required by 21 CFR 820.30(g). Specifically, your firm’s procedure “Health Hazard Evaluation”, Document # 701-105-546, has not been adequately implemented to evaluate health risk(s). Your firm’s procedure describes requirements for calculating health risk and conducting an analysis of complaint data. The determination of criteria for the complaint search and analysis does not ensure that all applicable or potential failure mode codes are selected.
Further,
The complaint search criteria used in your HHE do not encompass failure modes identified in your Risk Assessment and Controls Report that would be conservatively included and analyzed in your firm’s HHE risk assessment.
In this situation, the manufacture used only a subset of failure modes and harms to analyze complaints as part of the HHE risk of the polyethylene shoulder implants packaged in bags that do not meet the material specification requirement or oxygen transmission rate requirements. As a result, they missed many other potential failure modes that should have been considered in the analysis.
Key point: Clause 10 in ISO 1497111 requires manufacturers to establish a system to “actively collect and review information relevant to the medical device” that may affect safety. Complaints, including reports of adverse events, are one of the sources of information that must be analyzed, preferably using valid statistical methods. An effective post-market safety surveillance system is important for signal detection, risk reduction and creating a feedback channel for corrective and preventive action.
Disconnect #4: Ambiguous or insufficient definition of harm severity levels
In a warning letter issued to a leading manufacturer immunoassay analyzers and assays, FDA cites the following observation:
Failure to adequately establish and maintain procedures for risk analysis, as required by 21 CFR 820.30(g). Specifically, your firm's primary risk control procedure "Product Safety Risk Management," GLB-QS-PCD-0047, Revision 15.3, Dated 17Aug 2023, states that it is compliant with ISO 14971:2019 and EN 14971:2019 + A11:2021, and defines the (b)(4) severity of harm categories as: (b)(4). While the definitions for (b)(4) and (b)(4) as outlined in Appendix 9.1 are clear, the definitions of (b)(4), (b)(4), and (b)(4) are insufficient.
Since many relevant details have been redacted from this warning letter, it is difficult to understand the main issue in this case. So, let us break it down:
The risk control procedure has 5 different levels for severity of harm, consistent with current industry practice.
Definitions for two of these severity levels are clear, but the other three levels are not defined in sufficient detail, and possibly contain contradictory language.
The result of this ambiguity is that severity rating has been incorrectly selected for false result failure modes of assays that are used for making treatment decisions in various life-critical medical conditions such as Hepatitis B infection, heart attack and therapeutic drug monitoring.
Key point: ISO 14971 defines the term risk as the combination of the probability of occurrence of harm and the severity of that harm12. A common industry practice, consistent with guidance in ISO/TR 2497113 is to use 3 or 5 level scales for both probability and severity. If these levels are not defined in sufficient details, risk may be either under or over estimated because of incorrect assignment. Correctly estimating risk is critical for evaluating risk acceptability for considering risk control actions, preferably through design. An industry best practice is to use standardized terms and severity levels based on clinical experience for more accurate risk analysis.
Disconnect #5: Failing to connect CAPA actions with design controls
In a warning letter issued to a leading manufacturer of infusion systems14, FDA cites the following observation:
Specifically, your risk analysis is inadequate in that it was not updated as required per Risk Management Procedure, 410-0009-01, Revision: 17.
The Ivenix Large Volume Pump’s Software Failure Analysis, 550-0015-04, Rev 2.0, did not include the hazard of extended start-up time to achieve the stated flow rate accuracy of +/-5%, nor was it revised to include this hazard after becoming aware of a product defect that allowed the device to exhibit this issue when infusing fluids at a rate of >50 ml/hr. and <200 ml/hr., as documented and identified in CAPA-00038 and FAI-4446. CAPA-00038 was initiated on April 1, 2022, due to startup time failures in high flow of the Ivenix Large Volume Infusion Pumps. Furthermore, this quality issue resulted in the issuance of a customer notification on April 22, 2022, and ultimately the Class II recall RES 92973.
In a way, this disconnect is similar to disconnect #1 and #2 noted above. Here is another example of not updating the risk analysis when new product failures are observed. The main disconnect here is between the CAPA process and the risk management process, which seems to suggest that appropriate corrective and/or preventive actions were not taken through the design process.
The manufacturer responded by updating the risk analysis and training to affected personnel. However, FDA did not find it to be adequate:
Your responses are inadequate because they do not indicate that you plan to perform a retrospective review of CAPAs to ensure your risk analysis has been adequately updated, and there is no available evidence to document that your planned corrective actions will prevent recurrence of these violations.
Key point: Risk management activities span across multiple processes of the QMS. It is important to understand the flow of information in and out of these processes to ensure that risk analysis is adequately updated to drive appropriate actions. This is one reason why ISO 13485 requires a process approach15 to quality management. Risk analysis should not be treated as an isolated, independent activity; rather as an integral component of the QMS.
In conclusion
The disconnect between risk management activities and the design control process is a significant issue in the medical device industry. According to FDA inspectional observation data, 45% of findings in design validation are rooted in risk analysis.
FDA scrutiny on risk management activities, especially in the context of design and development, will only increase under the QMSR. This is because the concept of a risk-based approach to key processes of the quality system is a foundational aspect of ISO 13485:2016, which also outlines specific risk-based requirements for the QMS.
By studying recent warning letters, we can become aware of potential disconnects between risk management activities and design controls. These disconnects generally lead to not updating risk analysis, which is a key input to the design process. If the output of the risk management process is not flowing correctly in the design process, there is a chance that many known and potential risks are not adequately controlled. This disconnect has a serious consequence for continued safety and effectiveness of a medical device.
It is useful to take a careful look the risk management process, understand its inputs and outputs, and how they connect with other processes of the QMS, including the design and development process.
See 21 CFR 820 Subpart J - Corrective and Preventive Action
FDA: Inspection Observations, Updated November 22, 2023.
FDA: Data downloaded from FDA Inspections Dashboard, Accessed October 15, 2024.
See Federal Register, Vol. 61, N0. 195, Current Good Manufacturing Practice (CGMP) Final Rule; Quality System Regulation, issued Oct 7, 1996.
ISO 13485: Medical devices - Quality management systems - Requirements for regulatory purposes, 3rd edition, 2016.
ISO 14971: Medical devices - Application of risk management to medical devices, 3rd edition, 2019.
FDA: Warning letter issued to CardioQuip, LLC, CMS 621738, February 11, 2022
ISO 14971:2019 - Clause 4.5, Risk management file.
FDA: Warning letter issued to Criticare Technologies, Inc., CMS 686915, July 12, 2024.
FDA: Warning letter issued to Exactech, Inc., CMS 669904, January 19, 2024.
ISO 14971:2019 - Clause 10, Production and post-production activities.
ISO 14971:2019 - Clause 3, Terms and definitions. The term risk is defined in clause 3.18.
ISO/TR 24971: Medical devices - Guidance on the application of ISO 14971, 2nd edition, 2020.
FDA: Warning letter issued to Fresenius Kabi AG, CMS 671249, January 4, 2024.
ISO 13485: See Clause 0.3, Process approach, 3rd edition, 2016.
Excellent article with great insight to the FDA Observations. I might add more to the Risk Analysis section on the use of standards to identify Hazards and to provide Risk Controls and Risk Verifications of Effectiveness. This improves the efficiency of the risk management process. A second thought is that the ISO 13485:2016 7.3.3 c) citation on "the outputs of risk management" are design inputs, heightens the pressure on starting risk management BEFORE design input. Risk management should be started in feasibility taking the known standards that apply and using them as mentioned above, and any other knowledge of the proposed device in a tool such as PSA to start the risk management process. Risk Analysis is a requirement prior to conducting any clinical trials where a device is used on humans, is there is risk information very early in feasibility. This approach will reduce time and improve costs of product development over finding and fixing problems after the design is "complete", such as in Design Validation, or post-market complaints.